
Alpha-1-Antitrypsin-Mangel: blinder Fleck auch in der Pneumologie?
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Der Alpha-1-Antitrypsinmangel (AATM) gilt als seltene genetische Erkrankung und betrifft überwiegend die Lunge und die Leber,jedoch mithoher klinischer Variabilität. Doch AATM ist nicht so selten, wie wir annehmen, sondern häufig nur unterdiagnostiziert.
Keypoints
-
Unsere bisherige Praxis im Umgang mit dem AATM übersieht einen Großteil von Genveränderungen und damit von Menschen „im Risiko“.
-
In pneumologischen Kliniken und Praxen sollten (nahezu alle) Patienten in Bezug auf einen AATM evaluiert werden.
-
Je früher ein AATM erkannt wird, desto eher können Organschäden durch Lebensstilmodifikation, Monitoring und adäquate und frühzeitige Therapie vermieden werden („Team Prävention“).
-
Die aktuelle diskriminative Schwelle und auch das Ergebnis der Genotypisierung sind klinisch im Kontext zu werten und im Zweifelsfall ist eine Sequenzierung des SERPINA1-Gens durchzuführen.
Es muss davon ausgegangen werden, dass der Alpha-1-Antitrypsinmangel (AATM) häufig nicht diagnostiziert wird, da nicht gezielt danach gesucht wird. Damit wäre der AATM eine häufig nicht erkannte „seltene“ Erkrankung und die Prävalenz deutlich höher als bisher angenommen. Ein frühes Erkennen des AATM ist wichtig, um über Verhaltensmodifikation und Therapie irreversible Erkrankungen an Lunge und Leber verhindern zu können. Das Bild des typischen Alpha-1-Patienten –männlich, 50 Jahre alt, Raucher, Emphysem – muss als obsolet gelten.
Der Alpha-1-Antitrypsinmangel
Alpha-1-Antitrypsin (AAT) ist der wichtigste Proteasen-Inhibitor in der Lunge. Beim AATM kommt es durch genetische Veränderungen (>200 beschriebene Varianten)1 auf einem (heterozygote Form) oder beiden Alpha-1-Antitrypsin-Genen (homozygote oder „compound“-heterozygote Form,SERPINA1) zu einer falschen Zusammensetzung des AAT in den Leberzellen, dem Hauptproduktionsort. Die Folge ist eine geringere oder fehlende Ausschleusung des AAT oder die Freisetzung eines dysfunktionalen AAT in den Kreislauf. In den Leberzellen entsteht damit ein Überschuss an AAT mit der Folge des Zellfunktionsverlusts und -untergangs. Dies bedeutet eine Erhöhung des Risikos für die Ausbildung einer Leberfibrose (bei 20–36% der Patienten mit dem häufigsten Genotyp PI*ZZ)2, einer Leberzirrhose und auch eines Leberzellkrebses (HCC) ab ca. dem 50. Lebensjahr3.
Weitere Risikofaktoren wie männliches Geschlecht, Adipositas, Diabetes oder Alkoholkonsum tragen zur Erhöhung des Risikos für die Leber bei.2,3 Ein erster Gipfel bei der Erkrankung der Leber betrifft bereits Säuglinge. In einer schwedischen Kohorte wurden 120 von 200000 Neugeborenen (0,06%) mit dem homozygoten Typ PI*ZZ gefunden, wovon 12% eine prolongierte Gelbsucht (neonatale Cholestase) aufwiesen und 2–3% eine Lebertransplantation bei Leberversagen benötigten.4
Durch den Mangel an AAT im Blut und damit am Hauptwirkort, der Lunge, kommt es zu einemAusbleiben der antiproteolytischen Wirkung insbesondere gegenüber der neutrophilen Elastase (NE), wodurch die Zerstörung von Alveolarsepten und die Ausbildung eines Lungenemphysems begünstigt werden.5 Insbesondere das inhalative Rauchen stellt für Patienten mit einem AATM einen Hauptrisikofaktor für die Entwicklung eines (frühen, auch schon vor dem 40. Lebensjahr auftretenden)6 Lungenemphysems dar. AATM kann für ca. 1% der COPD-Fälle und für ca. 2% der Emphysemfälle ursächlich sein.5
Insgesamt zeigt der AATM eine hohe Variabilität in Bezug auf das klinische Erscheinungsbild von asymptomatisch homozygoten Genträgern bis zu heterozygoten (rauchenden) Genträgern mit schweren Erkrankungen.7 Der FEV1-Wert hat sich dabei als schlechter Surrogatparameter herausgestellt, bei AATM-Patienten sollte insbesondere die Diffusion untersucht werden.7
Aktuelle Prävalenzzahlen
Weltweit gibt es deutliche regionale Unterschiede in der AATM-Prävalenz.8 Die Variante M ist die Wildtyp-Variante („normal“). Die häufigsten klinisch relevanten Varianten weltweit sind S und Z (zwischen 1–5% der Bevölkerung; Tab. 1), gefolgt von den selteneren Varianten (<1% der Bevölkerung) F und I.1,3 In der ClinVar-Datenbank des National Center for Biotechnology Information (NCBI) sind mittlerweile weit mehr als 200 Varianten gelistet. Es wird jedoch selbst bei schweren AATM-Erkrankungen von einer Dunkelziffer von 90% ausgegangen.1,2
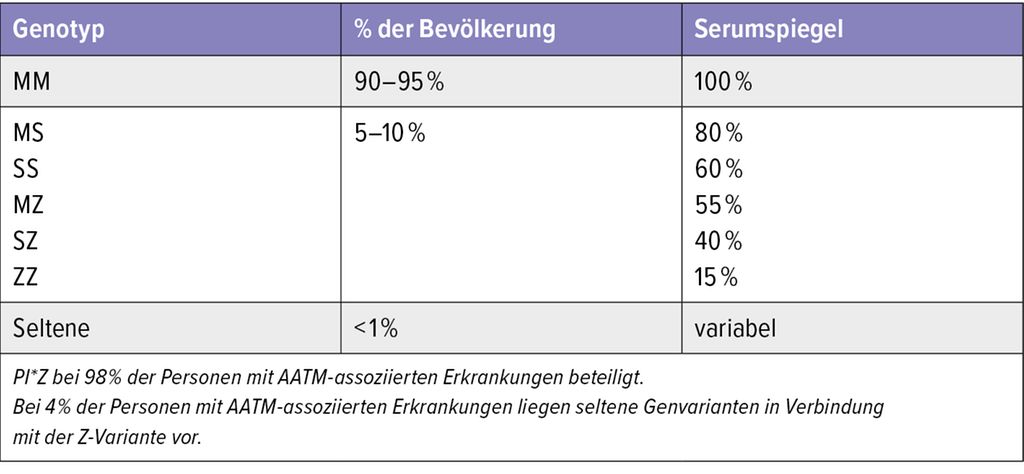
Tab. 1: Häufigkeit und Serumspiegel der häufigsten AAT-Genotypen (modifiziert nach Blanco I et al. 2017)8
Für Mitteleuropa (DE, AT, CH, PL) wird eine Häufigkeit des Z-Gens (PI*Z pro 1000 Einwohner) von 13 angegeben, für Nordeuropa von 20, für Westeuropa von 17, für Südeuropa von 17 und für Osteuropa von 88. Die Prävalenz des schweren AATM (PI*ZZ) und die Schätzung der Zahl der Betroffenen (in Klammer) für die entsprechenden Regionen liegen bei 1:5771 (Mitteleuropa; n=29903); 1:2041 (Nordeuropa; n=15553), 1:4992 (Westeuropa; n=41046),1:3785 (Südeuropa; n=30118) und 1:35702 (Osteuropa; n=2954).
International liegen die Zahlen für PI*Z in Nordamerika bei 12/1000, PI*ZZ-Prävalenz 1:13489 (n=73922), in Zentralamerika und der Karibik bei 5 bzw. 1:108252 (n=2781), in Südamerika bei 6 bzw. 1:26002 (n=14787), in Afrika bei 2 (n=3824 von > 1,2 Mrd. Einwohnern) und in Ost- und Südostasien bei 2 (n=10415 bei > 2 Mrd. Einwohnern).8 Es muss davon ausgegangen werden, dass diese Schätzungen nur einen Teil der Alpha-1-Realität abbilden und es eine deutlich höhere Dunkelziffer gibt.
Die Häufigkeit des Genotyps PI*MZ wurde für Europa mit 1:58 Einwohner angegeben (Mitteleuropa 1,1% der Bevölkerung), Amerika: 1:91, Afrika: 1:125, Asien: 1:337,Australien/Neuseeland: 1:329. In Bevölkerungsgruppen europäischer Abstammung liegt bei 2–4% ein MZ-Trägerstatus vor. In den untersuchten Regionen wird von einer Zahl von mehr als 35 Millionen MZ-Trägern ausgegangen. Die hohe Zahl an heterozygoten Genträgern in Europa erhält vor dem Hintergrund der weiterhin hohen Rauchquoten in den europäischen Ländern eine besondere Bedeutung, da rauchende MZ-Patienten ein erhöhtes Risiko für Lungenemphysem und COPD im Vergleich zu nicht rauchenden MZ-Patienten haben und auch das Leberrisiko9 bei erwachsenen MZ-Patienten erhöht zu sein scheint. Für beispielhafte Städte in Deutschland, Österreich und der Schweiz zeigt Tabelle 2 die möglichen Zahlen für PI*Z, PI-ZZ und PI-MZ.
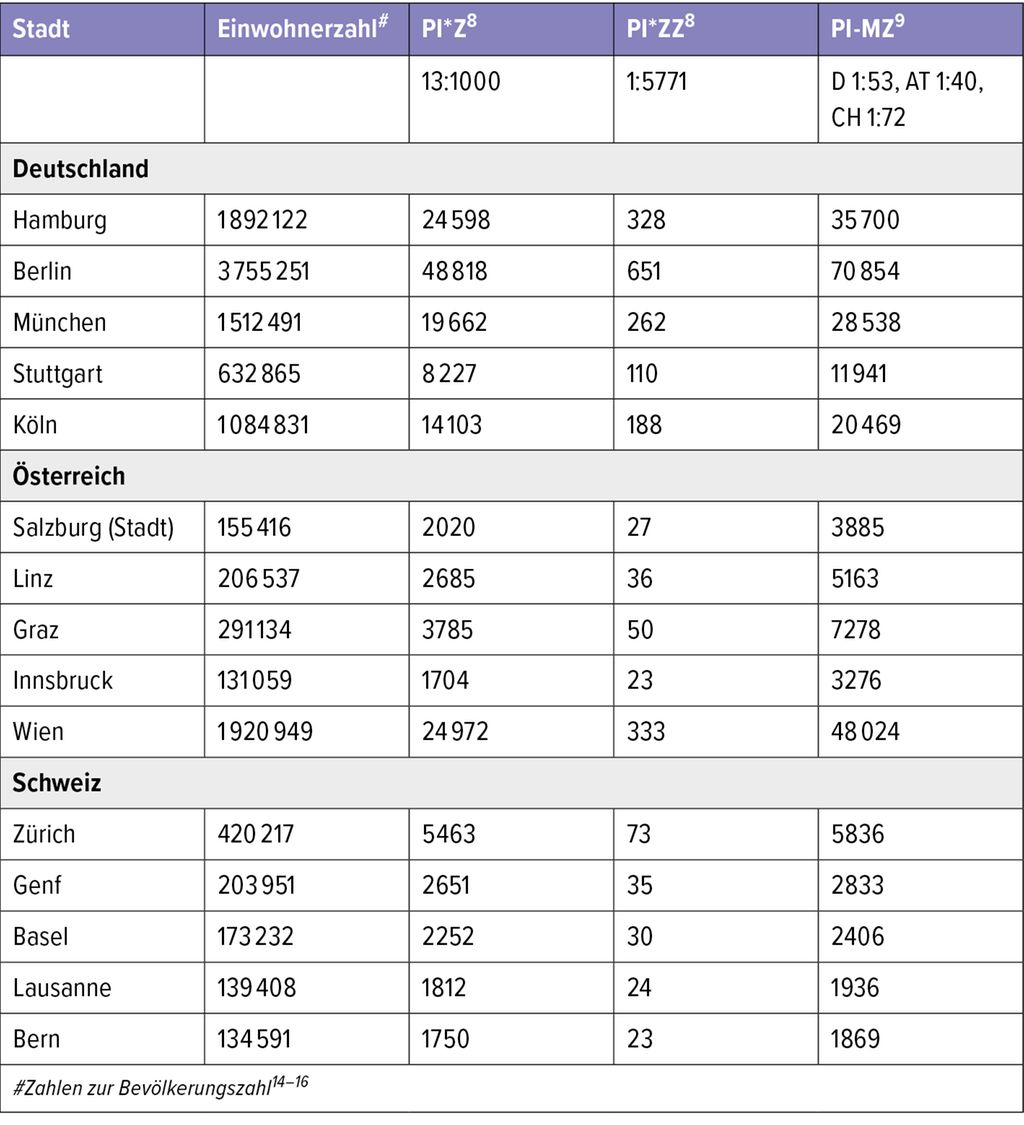
Tab. 2: Zahlen für PI*Z, PI-ZZ und PI-MZ für exemplarische Städte in Deutschland, Österreich und der Schweiz
Von wegen „männlich, 50 Jahre alt, Raucher, Emphysem“
In Deutschland gibt es derzeit unter dem Dach der Atemwegsliga e.V. 60 Alpha-1-Center für Erwachsene (42 Kliniken, 18 Praxen) und 10 für Kinder (ausschließlich Kliniken), deren Ziel eine bestmögliche Betreuung von Patienten mit AATM ist.10
Unsere eigene Praxis gehört zu den größeren Alpha-1-Zentren und betreut derzeit mehr als 300 Patienten mit AATM, 52 davon(17%) mit homozygotem Genstatus PI*ZZ, wovon 27 eine Augmentationstherapie erhalten. Pro Woche kommen in unserer Praxis im Mittel 1,6 neue Alpha-1-Patienten dazu – teilweise auf Zuweisung anderer Praxen zur Mitbetreuung, teilweise durch eigene Vorstellung von Patienten zur Zweitmeinung, die meisten jedoch im Zusammenhang mit einer aktiven Bestimmung des AAT-Serumspiegels bei jeder Blutabnahme in der Praxis, welche unabhängig von der Indikation der Blutabnahme erfolgt.
Von den errechneten Zahlen allein für die Stadt Stuttgart aus Tabelle 2 (11941 PI*MZ, 110 PI*ZZ) sind jedoch auch wir noch weit entfernt, was die Problematik der Dunkelziffer und der Unterdiagnose nochmals deutlich unterstreicht.
Das Engramm, das viele Kolleginnen und Kollegen mit dem AATM verbinden, ist ein Patient mit den Charakteristika „männlich, 50 Jahre alt, Raucher, Emphysem“. Dieses Bild ist einerseits falsch und trägt andererseits zur Unterdiagnose des AATM mit bei. In unserem Kollektiv sind 50% jünger als 52 Jahre (Altersmedian), 59% weiblichen Geschlechts und mehr als 55% haben eine Asthmadiagnose. Die „klassische“ Diagnose COPD/Emphysem liegt nur bei 15% unserer Alpha-1-Patienten vor (Abb. 1).
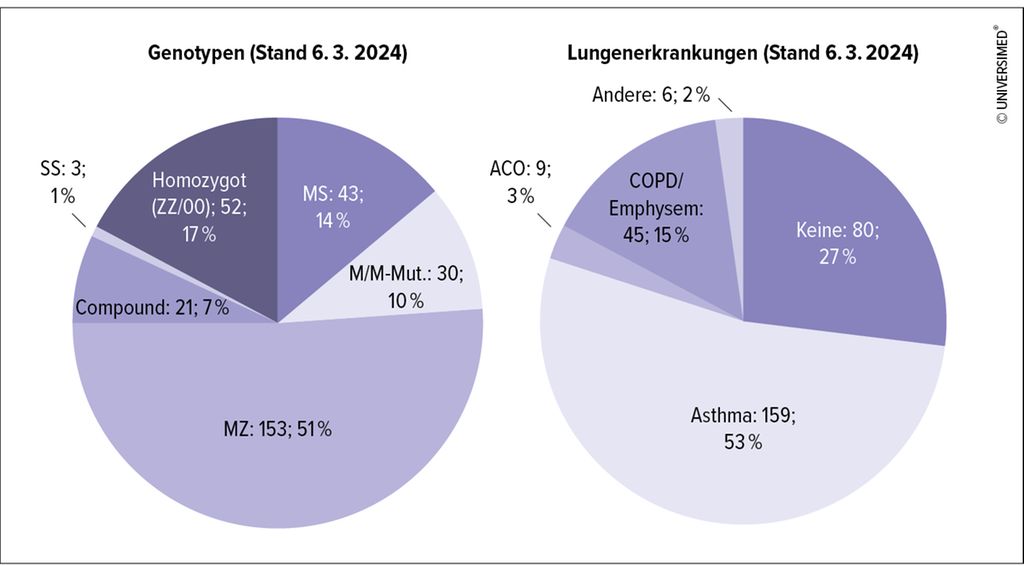
Abb. 1: Verteilung der Genotypen und der Lungenerkrankungen bei Alpha-1-Patienten des Alpha-1-Centers Pneumologische Praxis im Zentrum, Stuttgart; gesamt n = 30 (modifiziert nach Rupp A et al.)17
Abklärung auf AATM – Mission Prävention
Bei der Abklärung in Bezug auf einen AATM gibt es zwei unterschiedliche Herangehensweisen. Das „Team schwere Fälle“ möchte die Patienten mit Lungenerkrankungen aufgrund eines schweren Alpha-1-Antitrypsinmangels (i.d.R. PI*ZZ) identifizieren und sie einer wöchentlichen i.v.-Augmentationstherapie zuführen.
Das „Team Prävention“ möchte neben den schweren Mangelerkrankungen auch weniger gravierende Genveränderungen (wie z.B. PI*MZ) feststellen – idealerweise bevor irreversible Lungen- oder Leberschäden entstehen und diese Patienten „im Risiko“ durch Lebensstilmodifikation (Berufswahl, Nichtrauchen, Meidung/Reduktion pneumotoxischer oder hepatotoxischer Substanzen, Gewichtsregulierung, Bewegung, Asthma- und Allergiebehandlung), regelmäßiges Monitoring und adäquate Therapie optimal behandeln, um auch das Risiko für eine zukünftige Entstehung von Organschäden zu reduzieren. Auch bei heterozygoten Genträgern liegen durch koexistente genetische Faktoren oder durch Umwelteinflüsse ein Lungen- und Leberrisiko sowie ein Erkrankungsrisiko z.B. fürVaskulitis, Gallensteine oder COPD vor.2 So wurde eine erhöhte Odds-Ratio (OR) von 2,31 (1,60–3,35) für die Entwicklung von COPD von MZ-Patienten versus MM-Patienten gefunden.11
Wenn AATM erst dann diagnostiziert wird, wenn Lunge oder Leber Organschäden aufweisen, ist es bereits zu spät. Die „Mission“ unseres Alpha-1-Centers ist es, den AATM nicht erst zu erkennen, wenn die Patienten mit 47 Jahren aufgrund des inhalativen Rauchens bei homozygotem Mangel eine FEV1 von 24% vom Soll und eine CO-Diffusionskapazität (DLCO) von 41% vom Soll haben. Die Vorteile einer frühen Detektion des AATM überwiegen nach unserer Erfahrung potenzielle Nachteile (Tab. 3). Bei Vorliegen eines AATM sollten Familienangehörige, Partner und eigene Kinder in die (fach-)genetische Beratung mit aufgenommen und ggf. ebenfalls getestet werden.
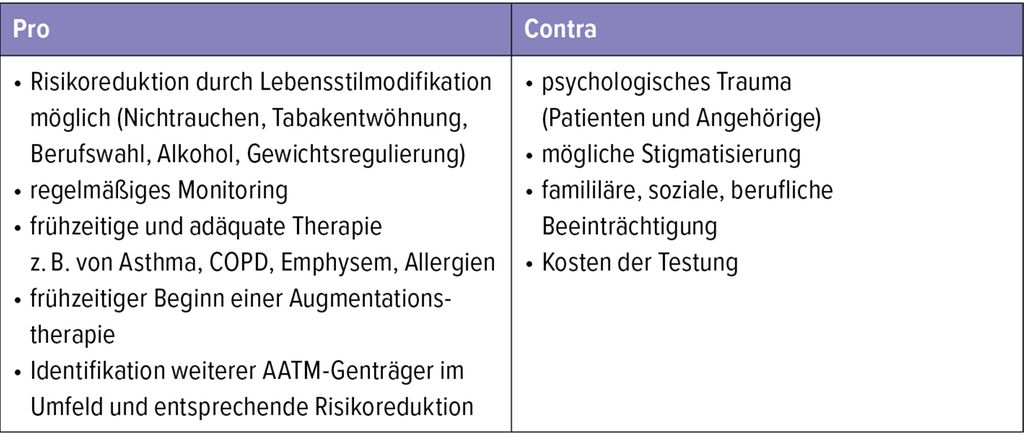
Tab. 3: Pro und Contra einer frühen Erkennung des AATM (modifiziert nach Miravitlles M et al. 2017 und Blanco I et al. 2017)7,8
Die Bedeutung der Prävention wird unterstrichen durch Zahlen der schwedischen Geburtskohorte, bei der in den Jahren 1972 bis 1974 200000 Neugeborene auf AATM gescreent wurden. 127 Neugeborene hatten den Genotyp PI*ZZ, 2 PI*ZQ0, 45 PI-SZ und 1 PI-SQ0. Die Kinder wussten somit von Geburt an über ihr Risiko Bescheid, dennoch hielt dieses Risikobewusstsein nicht alle davon ab, mit dem Rauchen zu beginnen. Bei einer Nachuntersuchung im Alter von 37 bis 40 Jahren waren 4% der ZZ- und SZ-Teilnehmer aktive Raucher und 18% bzw. 11% waren Ex-Raucher12 mit entsprechenden Einschränkungen der Lungenfunktionswerte bei den aktiven Rauchern, die einer COPD entsprechen (Tiffeneau-Index <70%). Eine Messung der Diffusion fand leider nicht statt. Auch in der Untersuchung von Holm et al. zeigten MZ- und SZ-Genträger höhere Raucherquoten als ZZ-Genträger und höhere Wahrscheinlichkeiten für ungesundes Verhalten wie Bewegungsmangel und Fehlgewichtigkeit.13
Pneumologische Patienten haben per se bereits ein höheres Risiko für einen AATM als Nichterkrankte oder Patienten in der Allgemeinmedizin. Daher sollten insbesondere Pneumologen bei ihren Patienten den AAT-Serumspiegel bestimmen. Bei einem Spiegel unterhalb der sog. diskriminativen Schwelle von 1,1g/l sollte eine Genotypisierung durchgeführt werden und bei unauffälligem Ergebnis und fortbestehendem klinischem Verdacht auf einen genetisch bedingten AATM auch eine Sequenzierung des SERPINA1-Gens.
Die diskriminative Schwelle von 1,1g/l mussnach unserem Dafürhalten insbesondere beijüngeren Patienten kritisch gesehen, werden, da 10% unserer Patienten mit bestätigtem AATM einen Serumspiegel >1,1g/l zeigten. Die Sensitivität der diskriminativen Schwelle liegt bei nur 73,4%, die Spezifität bei 88,5%.7 Auch die Genotypisierung (die in unserem Referenzlabor in Marburg 14 Genotypen beinhaltet und methodenbedingt Q0-Typen nicht erkennen kann) ist nicht der Weisheit letzter Schluss, da in 25% der Fälle, bei denen von uns bei unauffälliger Genotypisierung eine Sequenzierung nachgefordert worden war, ein positiver Befund mit einer „seltenen“ Genvariante gefunden werden konnte. Die klinische Relevanz seltener Genvarianten ist in vielen Fällen aufgrund der geringen Fallzahlen weltweit noch gar nicht klar, aber die Patienten können nach Diagnosestellung gemonitort werden und sollten in das europäische Register (EARCO) eingeschlossen werden, um longitudinale Beobachtungsdaten zu erhalten.
Literatur:
1 Ferrarotti I et al.: Rare variants in alpha 1 antitrypsin deficiency: a systematic literature review. Orphanet J Rare Dis 2024; 19(1): 82 2 Strnad P et al.: Alpha 1 antitrypsin deficiency. N Engl J Med 2020; 382(15): 1443-55 3 Fromme M et al.: Alpha-1 antitrypsin deficiency: A re-surfacing adult liver disorder. J Hepatol 2022; 76(4): 946-58 4 Sveger T: Liver disease in alpha 1-antitrypsin deficiency detected by screening of 200000 infants. N Engl J Med. 1976; 294(24): 1316-21 5 Mornex JF et al.: Alpha1-antitrypsin deficiency: an updated review. Presse Médicale. 2023; 52(3): 104170 6 Mostafavi B et al.: Lung function and CT lung densitometry in 37- to 39-year-old individuals with alpha-1-antitrypsin deficiency. Int J Chron Obstruct Pulmon Dis 2018; 13:3689-98 7 Miravitlles M et al.: European Respiratory Society statement: diagnosis and treatment of pulmonary disease in α 1 -antitrypsin deficiency. Eur Respir J 2017; 50(5): 1700610 8 Blanco I et al.: Alpha-1 antitrypsin Pi*Z gene frequency and Pi*ZZ genotype numbers worldwide: an update. Int J Chron Obstruct Pulmon Dis 2017; 12: 561-9 9 Martinez-González C et al.: Estimated prevalence and number of PiMZ genotypes of alpha-1 antitrypsin in seventy-four countries worldwide. Int J Chron Obstruct Pulmon Dis 2021; 16: 2617-30 10 Atemwegsliga: Alpha-1-Center in Deutschland. https://www.alpha-1-center.org/alpha-1-center.html ; zuletzt aufgerufen am 8.4.2024 11 Hersh CP: Chronic obstructive pulmonary disease in 1-antitrypsin PI MZ heterozygotes: a meta-analysis. Thorax 2004; 59(10): 843-9 12 Piitulainen Eet al.: Health status and lung function in the Swedish alpha 1-antitrypsin deficient cohort, identified by neonatal screening, at the age of 37–40 years. Int J Chron Obstruct Pulmon Dis 2017; 12: 495-500 13 Holm KE, Mannino DM, Choate R, Sandhaus RA: Genotype is associated with smoking and other key health behaviors among individuals with alpha-1 antitrypsin deficiency-associated lung disease. Respir Med 2018; 143: 48-55 14 Statistische Ämter des Bundes und der Länder. Gemeindeverzeichnis Deutschland 2022. https://www.statistikportal.de/de/gemeindeverzeichnis ; zuletzt aufgerufen am 8.4.2024 15 Statistik Austria: Gemeindeverzeichnis Österreich. https://www.statistik.at/fileadmin/publications/Gemeindeverzeichnis_Stand_1.1.2021.pdf; zuletzt aufgerufen am 8.4.2024 16 Bundesamt für Statistik Schweizerische Eidgenossenschaft: Regionalporträts 2021: Kennzahlen aller Gemeinden. https://www.bfs.admin.ch/bfs/de/home/statistiken/regionalstatistik/regionale-portraets-kennzahlen/gemeinden.assetdetail. 15864450.html ; zuletzt aufgerufen am 8.4.2024 17 Rupp A et al.: Charakteristika der Alpha1-Antitrypsinpatienten am Alpha1-Center Pneumologische Praxis im Zentrum, Stuttgart; eigene Daten. Stand 6.3.2024
Das könnte Sie auch interessieren:

Long-Covid-Rehabilitation 2025 – von der klinischen Erfahrung zur Evidenz
Seit 2020 hat sich die Rehabilitation bei Covid-19 und Long Covid von ersten Erfahrungsberichten hin zu einer evidenzbasierten Maßnahme entwickelt. Aktuelle Studien belegen, dass ...
Asthma bronchiale: Herausforderungen bei Kindern und Jugendlichen
Asthma bronchiale ist eine chronische Krankheit, die im Kindesalter andere Herausforderungen mit sich bringt als bei Erwachsenen. Die physikalische Untersuchung sowie die Therapie sind ...
